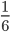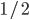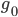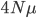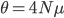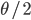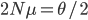A year or two ago I wrote a letter of recommendation for grad school for a former student of my genetics class. Last week they wrote me back and told me about their climate change manuscript (link) that was accepted for publication in Nature. The publication was also picked up by the media.
Here is the message:
How are things? I just wanted to say thanks again for everything that you did to help me get into grad school (you wrote me a letter of recommendation your first semester teaching genetics at UH fall of 2011). I wanted to thank you by sending you a copy of a paper that I was co-author on that just got published in Nature "The projected timing of climate departure". You may or may not have heard about this yet, but so far it is causing quite a media frenzy. I am sure that you will find this paper interesting as it has a heavy biodiversity focus and assesses the impacts of climate change in a new light, not just focusing on absolute changes. Anyways, I hope things are going well, and I just wanted to say thank you for everything and that you helped make a part of this.
Cheers,"
It feels wonderful to get this kind of positive feedback from students from time to time. In everyday teaching of a class it often feels like everyone is unhappy because you tend to only hear from the small fraction of students that complain.
Last Friday the lead author, Dr. Camilo Mora, gave a presentation on the work in our joint department seminar series. Most climate change predictions focus on the dramatic increase in temperature in the polar regions--and indeed this is where the largest temperature changes are expected to occur. But this is not the full picture. One question is how long until we will experience unprecedented change that is outside the range of previous temperatures. It turns out that this is predicted to happen first in the tropics, because the tropics have such stable temperatures, a smaller change can push the climate outside of the normal historical boundaries. The problem is that tropical species and environments cannot handle temperature changes as well as temperate and polar species, because they have not had to adapt to wide swings of temperature in the past. Another problem is that tropical countries tend to have less economic resources to deal with these changes. The predictions for "climate departure" for some of the tropics are surprisingly near with some dates as soon as 2020.
Dr. Mora was able to do this as a student class project and with very little resources available, using a lot of free tools and data available online.
Here are some news links:
http://www.latimes.com/nation/la-sci-climate-change-tropics-20131010,0,482935.story
http://www.popsci.com/article/science/no-year-after-2047-will-be-cool-now
http://www.weather.com/news/climate/2047-coldest-years-may-be-warmer-hottest-past-20131009
http://www.cbc.ca/news/canada/north/world-temperatures-go-off-the-chart-by-2047-study-says-1.2053488
http://www.sciencedaily.com/releases/2013/10/131009133216.htm